

Automating predictions of cancer patient outcomes

Jeffrey Spitzner, PhD1; Trevor Allen, BA1; Guy Jacks, BA1;James L Chen, MD1,2
1MatchTX, LLC, Columbus, Ohio. 2Dept of Biomedical Informatics and Internal Medicine, The Ohio State University, Columbus, Ohio
Poster was presented at BioIT World 2017, Boston MA
Background
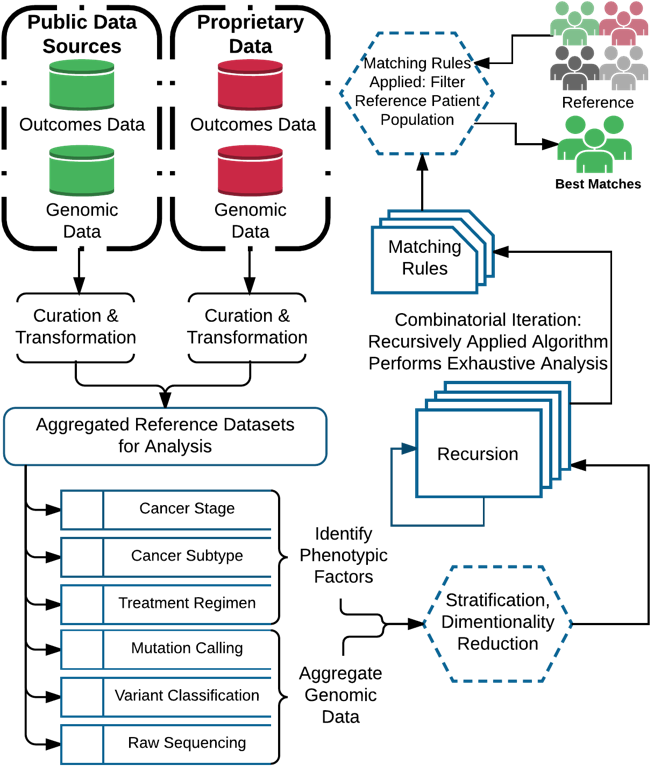
The early promise of whole genome sequencing and the reduction of molecular variants into clinically actionable diagnostics have been more difficult than initially anticipated. The direct translation of small subsets of variants has not encompassed the required complex nature to interpret heterogeneous diseases, and the algorithms that call these variants are not yet optimized. An alternative to this approach is to focus on the use of complex feature sets that include clinicogenomic features to match patients to patients and then extrapolate information about the disease from the matching patients. We introduce a solution MatchTX, which combines genomics data outcomes data for conducting patient cohort matching.
Methods
Patient data: Clinical outcomes and molecular next-generation sequencing (NGS) data were obtained from The Cancer Genome Atlas (TCGA) and from deidentified patient datasets from OSU.
Network construction: Clinical outcomes and NGS data from TCGA are integrated to generate a predictive outcomes network (PON). Patients are compared to other patients within the PON to calculate network distances based on their genomic profiles. Patients with the smallest distances are determined to be best matches. Best match cohorts are then used to infer outcomes for unknown patients.
Development: MatchTX is developed in Django, a Python web framework with data currently sored in Postgres. Computational analysis is performed in Python.
Results
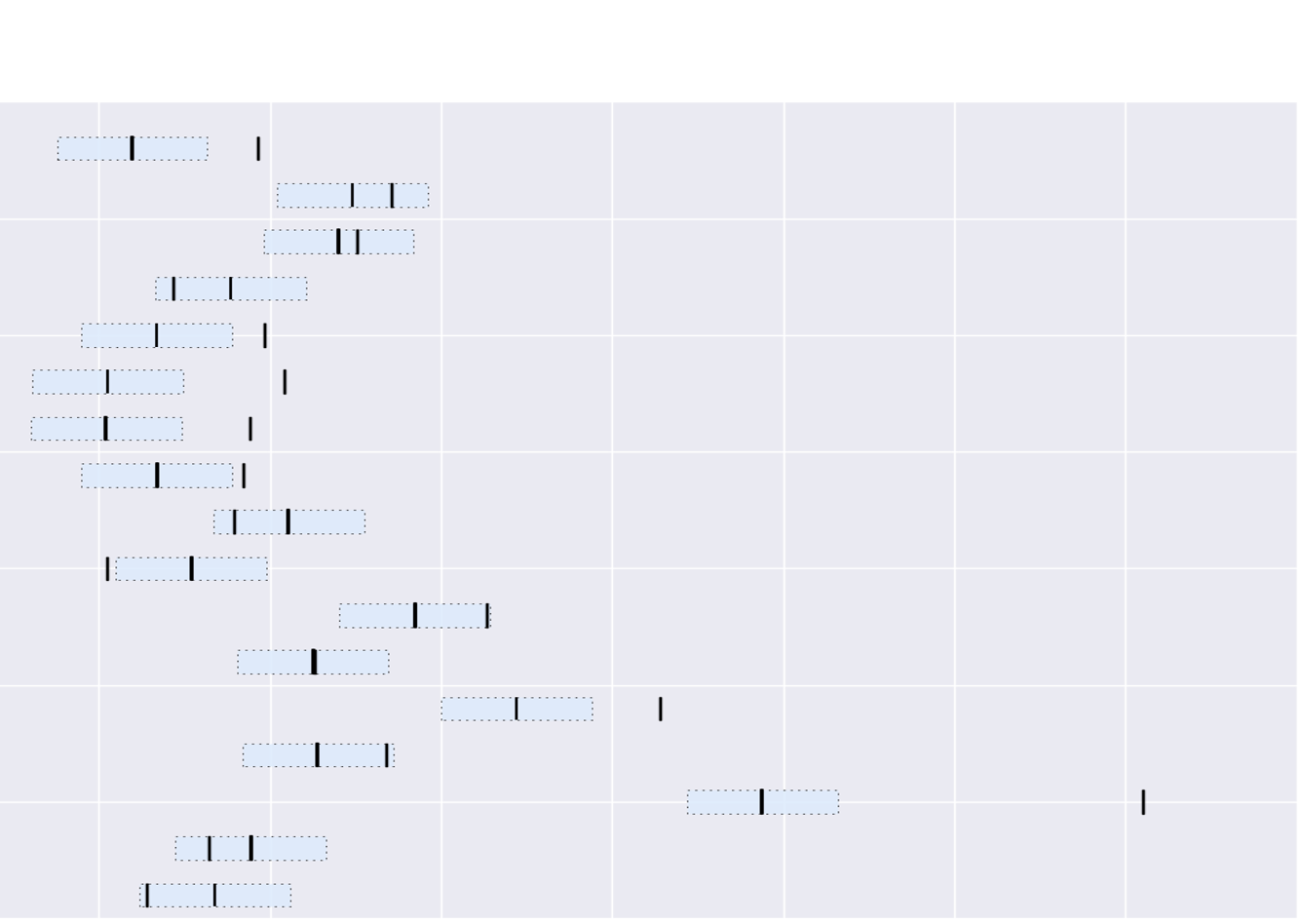
Reference Set: 130 patients with gallbladder cancer seen at The Ohio State
Test Set: 17 patients obtained from The Cancer Genome Atlas
Matching Rules: 272,676 (nom p-value 0.01)
Predictive Accuracy: The date of death was predicted within 0.5SD in 10 patients correctly.
Conclusion
Integrated genomic cohort matching through MatchTX provides an automated approach to predicting outcome in cancer patients.
MatchTX may be applied to different areas of clinical medicine. More information can be found at www.match-tx.com.

DEPARTMENT OF BIOMEDICAL INFORMATICS, OSU